Von-Hippel-Lindau-Syndrom (VHLS)
Die von Hippel-Lindau Erkrankung ist eine seltene, erbliche Tumorerkrankung. Insbesondere können Tumoren (Angiome) auftreten auf der Netzhaut und in Kleinhirn und Rückenmark sowie Tumoren in der Niere, Nebenniere und der Bauchspeicheldrüse. Zysten treten häufig in Niere und Bauchspeicheldrüse auf.
Definition und Krankheitsbild:
Die Von-Hippel-Lindau´sche Krankheit gehört zu den sogenannten Phakomatosen. Es handelt sich um eine seltene erbliche Tumorerkrankung verschiedener Organe, bei der Männer und Frauen gleichermaßen betroffen sein können. Am VHL-Syndrom erkranken in Deutschland jährlich etwa 2500 Personen.
Vererbung und Genetik:
Die Vererbung erfolgt autosomal-dominant mit unterschiedlicher Penetranz. Das bedeutet, dass die Anlage auf 50% der Kinder übergeht, wobei nicht jedes dieser Kinder erkranken muss. Hat ein Kind keine Anlage, kann es weder das VHL-Syndrom übertragen noch daran erkranken. Die Genveränderung liegt auf dem Chromosom 3p25-26 (VHL-Gen). Durch eine genetische Untersuchung von Betroffenen und Familienangehörigen kann festgestellt werden, ob eine Anlageträgerschaft vorliegt.
Das Auftreten von typischen klinischen Symptomen, beziehungsweise die Diagnosestellung ist zeitlich sehr unterschiedlich, ebenso wie der Schweregrad der Erkrankung. Die Erkrankung kann durchaus auch schon in der Kindheit oder auch erst im Alter auffällig werden. Das mittlere Alter bei der Diagnosestellung liegt bei 20 bis 25 Jahren. Eine oder mehrere klinische Erkrankungen sind meist vor dem 30. Lebensjahr vorhanden. Eine Augenbeteiligung (Kapilläres Netzhaut-Hämangiom) tritt oft als Erstmanifestation auf, da hier Krankheitszeichen frühzeitig entstehen beziehungsweise bemerkt werden.
Das Krankheitsbild kann typischerweise folgende Organe umfassen:
- Augen
- Gehirn und Rückenmark
- Niere und Nebennniere
- Bauchspeicheldrüse
- Geschlechtsorgane
- Innenohr
Klinische Beschwerden und Komplikationen:
Augen: Kapilläres retinales Hämangiom (Netzhaut-Gefäßtumor)
Dieser Tumor findet sich bei 50 bis 60% aller Patienten mit VHLS, von Betroffenen haben die Hälfte mehrere und beidäugige Tumoren. Andere Bezeichnungen für diese Tumore sind Angiomatosis retinae oder Von-Hippel-Erkrankung. Netzhaut-Hämangiome werden meist im 2. Bis 3. Lebensjahrzehnt klinisch auffällig. Es handelt sich um gutartige kleine grau-rötliche Tumore der Netzhaut, seltener des Sehnervenkopfes, die sich häufig vorwölben und bei Größenzunahme typische Veränderungen der Netzhautgefäße verursachen (starke Erweiterung der zuführenden Schlagadern und abführenden Venen). Diese Tumore können einzeln oder vielzählig, ein- oder beidseitig auftreten. Einseitige und einzelne Tumore können auch bei sonst gesunden Personen ohne VHL-Syndrom auftreten, wobei bei beidseitigem oder vielzähligem Auftreten von Netzhaut-Hämangiomen immer eine ausführliche Untersuchung anderer Organsysteme durchgeführt werden sollte. Auch beschwerdefreie Angehörige dieser Patienten sollten augenärztlich untersucht werden. Es kann durch Komplikationen wie Austritt von Flüssigkeit und Fettablagerungen unter oder in die Netzhaut, besonders im Bereich der Stelle des schärfsten Sehens (Makula), zu einer Verschlechterung des Sehvermögens, einem Gesichtsfeldausfall sogar bis zur Erblindung des betroffenen Auges kommen.
Die Netzhauttumoren haben die Neigung zum Wachstum und können ferner zur Netzhautablösung, auch mit massiven Vernarbungen, zur Blutung in den Glaskörper, zum grauen Star (Linsentrübung, Cataract) und zum grünen Star durch Gefäßwucherungen (neovaskuläres Sekundärglaukom) und auch zur Zerstörung des Augen mit Schmerzen (Phthisis bulbi) führen. Die Möglichkeiten zur Therapie und die Prognose sind wesentlich abhängig von der Größe, Lage und bereits bestehenden Komplikationen zum Zeitpunkt der Diagnosestellung.
Diagnostisches Vorgehen / Untersuchungsmethoden:
Durch eine Spiegelung des Augenhintergrundes bei weitgestellter Pupille ist es möglich, Netzhauttumore zu erkennen. Zur genaueren Einordnung der Erkrankung ist es notwendig, weitere Untersuchungen in einer spezialisierten Klinik durchführen zu lassen. Besondere Bedeutung kommt der Ultraschalluntersuchung des Auges und der Fluoreszenz-Angiographie zu. Bei letzterer wird ein fluoreszeiernder Farbstoff in eine Vene gespritzt und mittels einer Spezialkamera die Blutversorgung und andere Besonderheiten des Netzhauttumors untersucht.
Therapie:
Ziel der Therapie der Netzhaut-Hämangiome muss die Erhaltung des Sehvermögens sein. Eine Photokoagulation, die Zerstörung des Tumors durch einen Laser, ist bei kleineren Tumoren (bis etwa 3 Millimeter Höhe) möglich, oft sind mehrere Therapiesitzungen notwendig, zumal die Netzhauttumoren zu Rezidiven (Auftreten neuer Tumoren bzw. erneutes Wachstum) neigen. Eine Kryokoagulation (Kältebehandlung) dient ebenfalls der Zerstörung der Tumoren und wird bei größeren Hämangiomen oder beim Auftreten der Tumoren in der Netzhautperipherie (von hinteren Pol entfernte Anteile des Netzhaut) angewandt. Auch hier sind oftmals mehrere Sitzungen der Therapie notwendig. Eine Kryokoagulation wird auch beim sogenannten Sekundärglaukom (grüner Star durch Gefäßwucherungen im Kammerwinkel), einer Komplikation der Netzhaut-Hämangiome, durchgeführt. Selten werden Diathermiekoagulation oder mikrochirurgische Tumorresektion angewandt. Bewährt hat sich bei fortgeschrittenen oder großen Hämangiomen oder bei starkem Flüssigkeitsaustritt aus dem Tumor die fokale Bestrahlung. Meist wird diese als Brachytherapie (Kontaktbestrahlung) mit einem operativ auf den Augapfel aufgebrachten Strahlenträger (Applikator) durchgeführt, seltener kommt die aufwändige Protonenbestrahlung bei sehnervennahen Tumoren zur Anwendung. Bei Komplikationen wie Netzhautablösung oder Blutung in den Glaskörper ist eine Vitrektomie (Glaskörperentfernung) notwendig, hier ist die Prognose für das Sehvermögen oftmals sehr schlecht. Eine Enukleation (Entfernung des Augapfels) kann selten bei fortgeschrittenem grünen Star oder Augapfelschrumpfung (Phthisis bulbi) und Schmerzen notwendig sein.
Gehirn und Rückenmark: Hämangioblastom (Gefäßtumor)
Hämangioblastome finden sich typischerweise im Kleinhirn, bei 40% der Patienten über dem 30. Lebensjahr und bei 70% der Patienten über dem 70. Lebensjahr. Im Rückenmark oder im Hirnstamm findet sich dieser Tumor bei 10 bis 15% der Patienten. Gefäßtumore sowohl im Gehirn als auch im Auge finden sich bei über 80% aller Personen mit VHLS. Hämangioblastome des Kleinhirns können Kopfschmerzen, Übelkeit und Gangunsicherheit bewirken. Tumoren im Rückenmark verursachen z.B. Taubheit in umschriebenen Hautbezirken und Schwäche einzelner Muskelgruppen. Zum Ausschluss eines Gehirn- oder Rückenmark-Tumors wird eine Kernspin-Tomographie oder eine Computertomographie (beides Schnittbildverfahren) mit Kontrastmittel durchgeführt.
Niere und Nebenniere:
Ein Nierenzell-Karzinom (Nierenkrebs) wird bei 5% aller über 30-jährigen Patienten und bei 40% der über 60-jährigen Patienten mit VHLS gefunden. In ¾ der Fällen sind beide Nieren erkrankt. Dieser Tumor ist bösartig und kann zu Tochtergeschwülsten (Metastasen) in andere Organen führen. Krankheitszeichen werden oftmals erst sehr spät als Ausdruck von Absiedlungen in Lunge, Leber, Knochen oder Gehirn erkannt. Ein Phäochromozytom (Adrenalin-bildender Tumor) ist ein bösartiger, seltener Tumor der Nebennieren. Durch Produktion des “Stresshormons” Adrenalin kann es zu hohem Blutdruck, Schweißausbrüchen, Kopfschmerzen und Pulsrasen kommen. Auch dieser Tumor kann in andere Organe streuen. Im Urin Betroffener lässt sich Vanillinmandelsäure, ein Abbauprodukt des Adrenalins, nachweisen. Nierenzysten (Flüssigkeitsgefüllte Hohlräume der Nieren) können ebenfalls beim VHLS auftreten und führen meist nicht zu Beschwerden oder Organschäden. Zur Untersuchung von Tumoren der Nieren (auch anderer Organe des Bauchraumes wie der Bauchspeicheldrüse) dient die Ultraschalluntersuchung, gegebenenfalls muss eine Computertomographie ergänzt werden.
Bauchspeicheldrüse:
Hier finden sich typischerweise Zysten, welche meist keine Beschwerden verursachen. Wesentlich seltener ist das Inselzell-Karzinom (Krebs der Insulin-produzierenden Zellen). Dieser Tumor kann durch Beeinflussung des Zuckerstoffwechsels zu gefährlichen Stoffwechselentgleisungen führen.
Geschlechtsorgane:
Nicht selten, meist jedoch ohne Krankheitszeichen, sind Zysten bei Männern in den Nebenhoden, bei Frauen in den Eierstöcken. Gutartige Tumoren der Nebenhoden (Zystadenome) sind selten, können jedoch Grund von Unfruchtbarkeit sein.
Ohren:
Äußerst selten sind Tumore des Innenohres, welche zu Taubheit führen können.
Richtlinien zur Vorsorgeuntersuchung beschwerdefreier Personen zur Diagnose von Erkrankungen bei VHL-Syndrom
Betroffene Patienten:
Jährlich: Körperliche Untersuchung (auch urologisch / gynäkologisch) Untersuchung des Augenhintergrundes Ultraschall der Bauchorgane (Nieren und Bauchspeicheldrüse) Untersuchung des 24-Stunden-Sammel-Urins auf Vanillinmandelsäure
Alle 3 Jahre: eventuell Computer-Tomographie der Bauchorgane Kernspin-Tomographie oder Computer-Tomographie von Gehirn und Rückenmark bei Personen unter dem 50. Lebensjahr
Alle 5 Jahre: Kernspin-Tomographie oder Computer-Tomographie von Gehirn und Rückenmark bei Personen über dem 50. Lebensjahr Risiko-Angehörige:
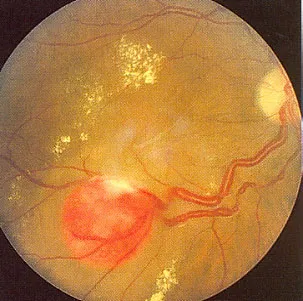
Abbildung 1: Typischer Befund eines Netzhaut-Hämangioms (am unteren Bildrand) mit Flüssigkeitsaustritt am hinteren Augenpol, Lipidexsudaten (gelblich) und stark erweiterten zu- und abführenden Gefäßen, rechts am Bildrand die Sehnervenpapille.
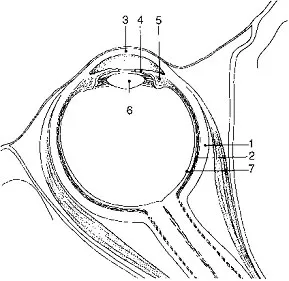
Abbildung 2: Querschnitts-Zeichung des Auges:
1 Lederhaut, 2 Aderhaut, 3 Hornhaut, 4 Regenbogenhaut, 5 Strahlenkörper, 6 Linse, 7 Netzhaut
Selbsthilfegruppe
Gerhard Alsmeier
Verein für von der Von-Hippel-Lindau-Erkrankung betroffenen Familien e.V.
Rembrandtstraße 2
49716 Meppen
Telefon: 05931-929552
Email: ed.uadnil-leppih@reiemsla.g
Website: www.hippel-lindau.de
Autor
Dr. med. Oliver Vij
Universitäts-Augenklinik Essen Tumorsprechstunde
Hufelandstr. 55
45122 Essen
Telefon: 0201-723-2900
Fax: 0201-723-5917
Email: ed.nesse-inu@jiv.revilo und ed.nesse-inu@ednutshcerpsromut
Website: www.uni-essen.de/augenklinik


